Cell Ranger2.1, printed on 04/23/2025
Chain View allows you to explore clonotype chain sequences at a contig-by-contig, base-by-base level. To enable Chain View, click on any chain in the clonotype list. Let's start by looking at the beta chain of the fourth clonotype in the clonotype list (TRB:TRBV4-2:TRBD1:TRBJ2-1:TRBC2; CDR3 CASSPRTGMDGNEQFF):
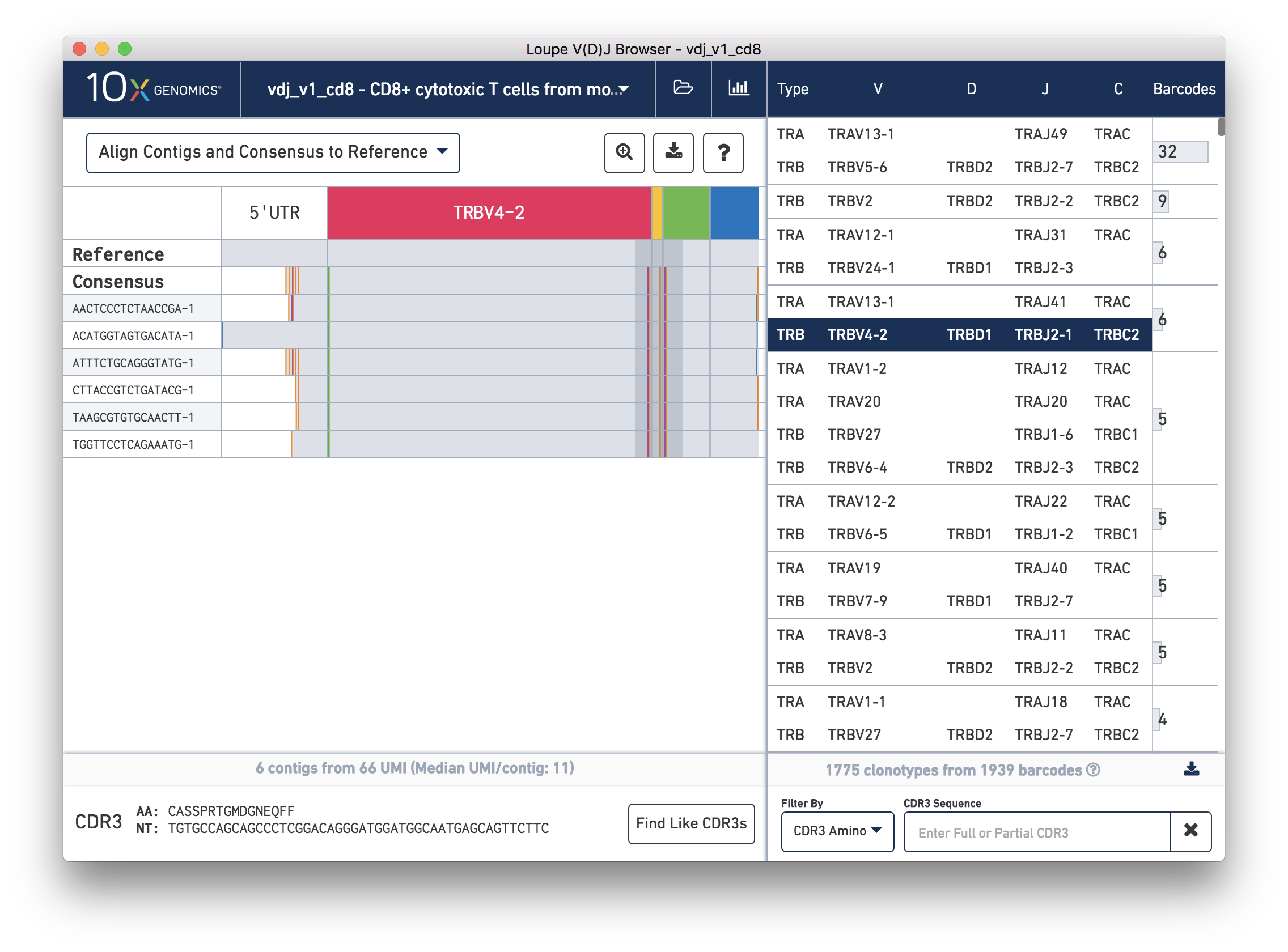
When you first click on a clonotype chain, you'll see a series of rows – grey bars of various widths preceded by a barcode label. The row marked Consensus above the contigs is the chain consensus sequence, a sequence that represents the most likely sequence for a receptor chain within the clonotype. Above the consensus is the Reference sequence, which is a concatenation of the reference sequences of each gene detected in the chain. Finally, atop the reference and consensus sequences are the gene annotations themselves, which indicate where the detected gene segments start and end. You can move your mouse over these annotations to see the gene name, or refer to the highlighted chain in the clonotype list.
As mentioned above, each row in this list represents a contig sequence. The gray backgrounds indicate where the contig sequences align to the reference. As you can see, the 5′ end of some of the rows are white, indicating that the assembled contig did not span the entire 5′ UTR region of the reference.
You can also see a series of vertical lines or blocks of various colors across some of the contigs. Each indicates a feature of interest, such as a start or stop codon or a variation from the reference. The button showing a question mark will display a legend showing what feature each color represents:
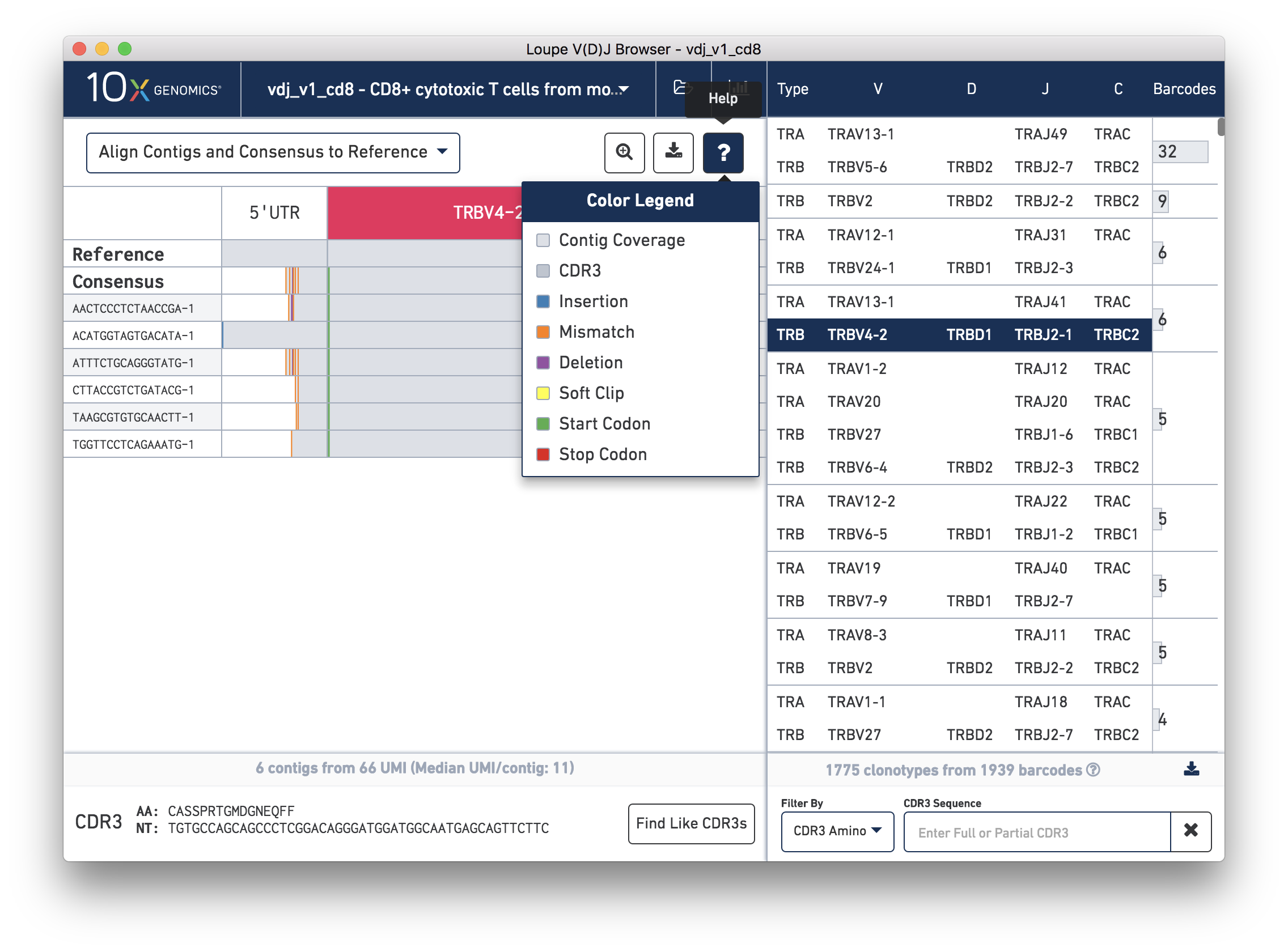
Usually, start codons appear at the very beginning of the V gene annotation, and stop codons will not be seen as they occur at the 3′ end of the C gene annotation downstream of where the primers amplify.
Somatic cells are not just characterized by their V, D, and J genes; the addition and deletion of bases at gene junctions during V(D)J recombination further contributes to lymphocyte diversity. Because of this additional variability, areas around gene junctions will often appear to have many mutations compared to a canonical reference, as you see in this chain. Note, however, that the consensus sequence has the same variants here as the contigs, because those variations are incorporated into the CDR3 that defines the clonotype.
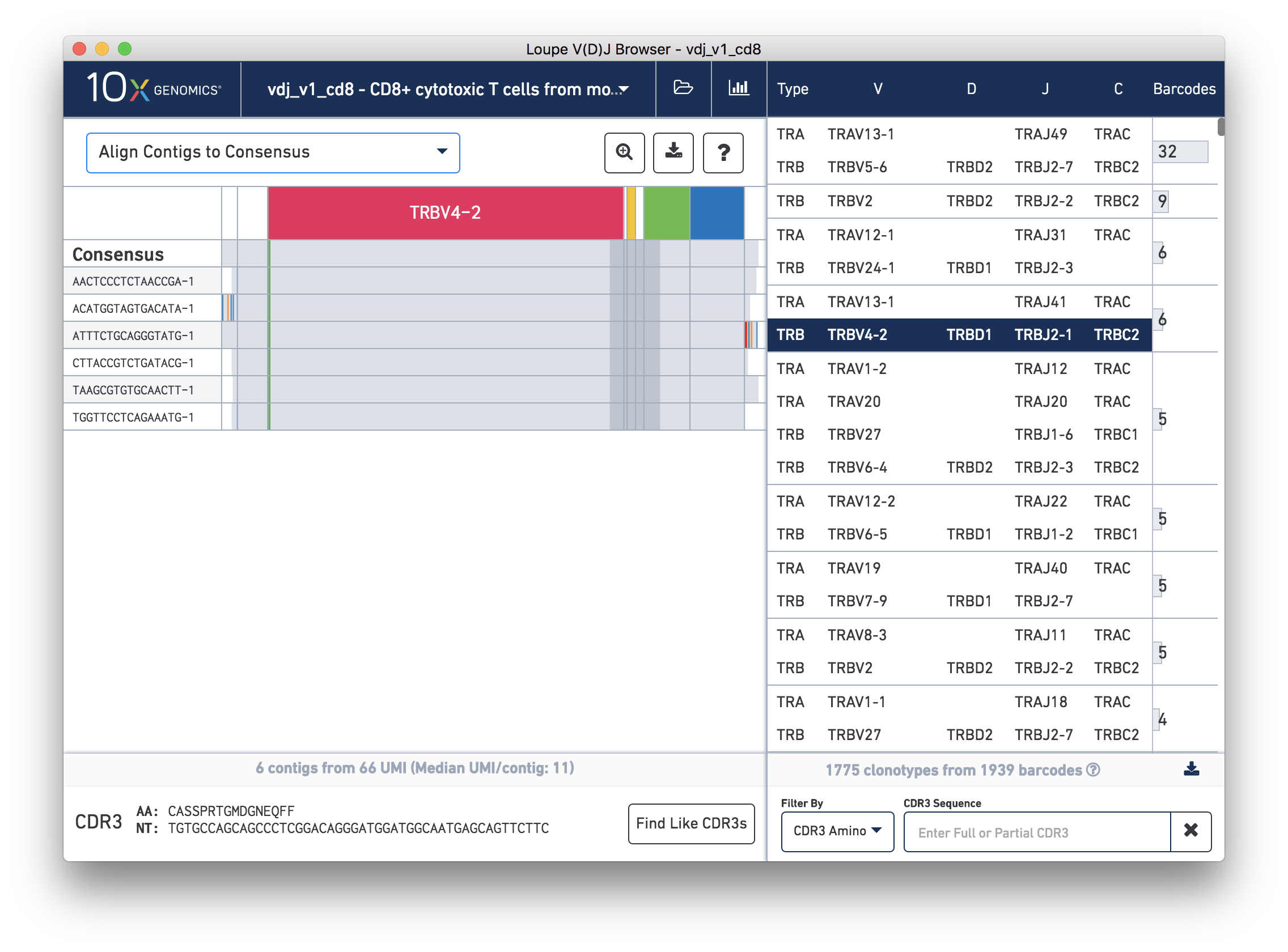
While it is useful to see the common mutations around gene junctions, they make it difficult to assess how closely the contigs map to the consensus sequence. You can more easily do this by visually aligning the contigs to the shared consensus sequence. To do so, click the selector above the gene annotations and select Align Contigs to Consensus.
Switching to consensus alignment view hides common intra-junction variability. It also shows additional gaps within these junctions, which correspond to non-template insertions. Outside the annotated regions are 5′ and 3′ unannotated regions. These regions may contain a many mismatches and technical artifacts. In the 3′ region of the third contig (ATTTCTGCAGGGTATG-1), you can see what is likely an errant stop codon.
Curious about the actual contig sequences? Let's take a look at another chain and explore the sequence level view.
We'll navigate to a more interesting chain to inspect at the base level. Click on the alpha chain of the same clonotype (TRA:TRAV13-1:TRAJ41:TRAC), and select Align Contigs and Consensus to Reference view in the top left dropdown.
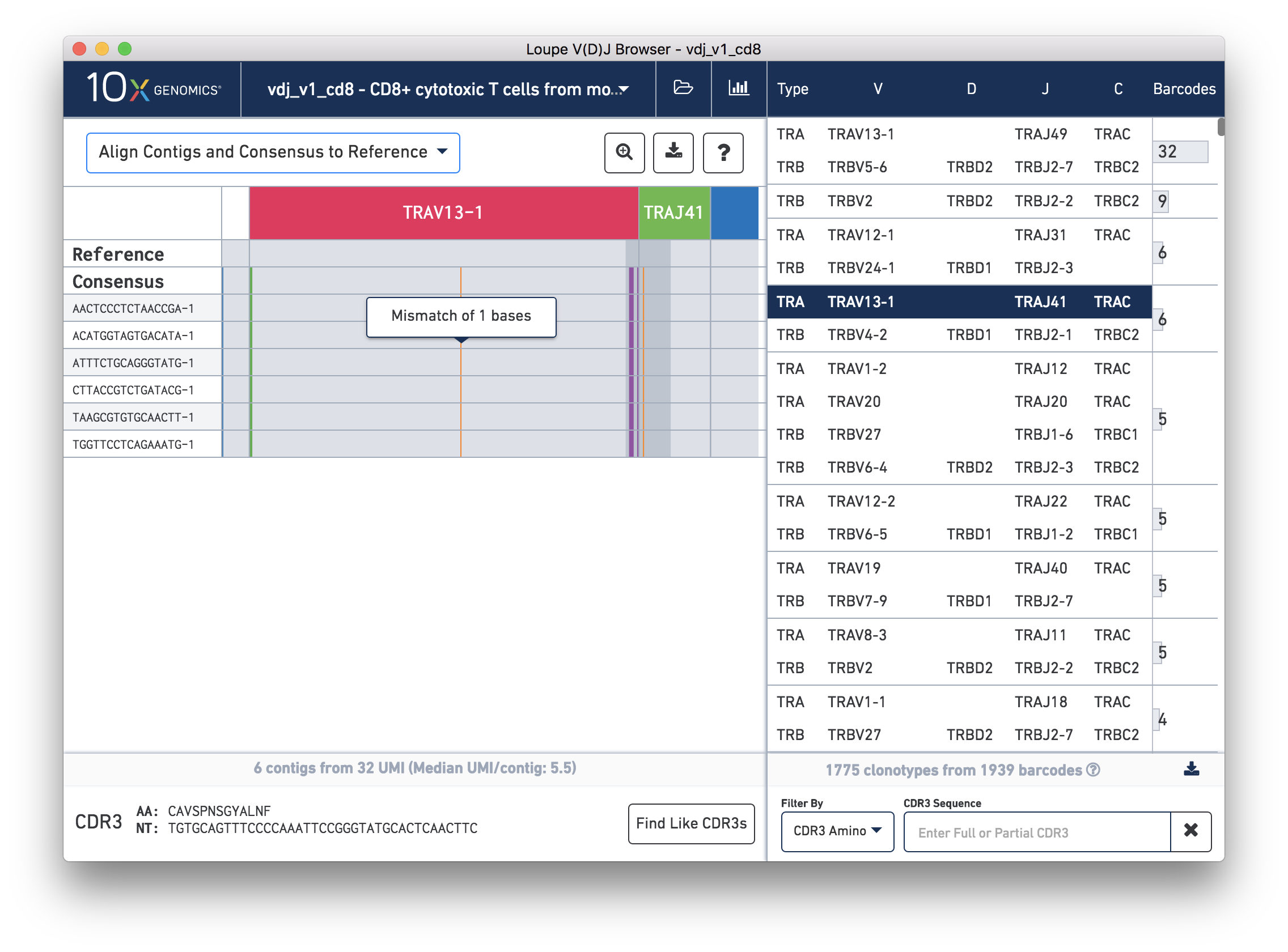
You're probably wondering what the single (orange) mismatch in the middle of the TRAV13-1 annotation is. Click on it to zoom in to the sequence level view and find out. After zooming in, the feature colors remain the same; orange still signifies mismatches. It looks like a single vertical row of bases are marked as mismatches. The reference base at this location is a G while our assembled contigs as well as the consensus at this location show an A. This SNP is listed in dbSNP. You can also view it in the UCSC Genome Browser.
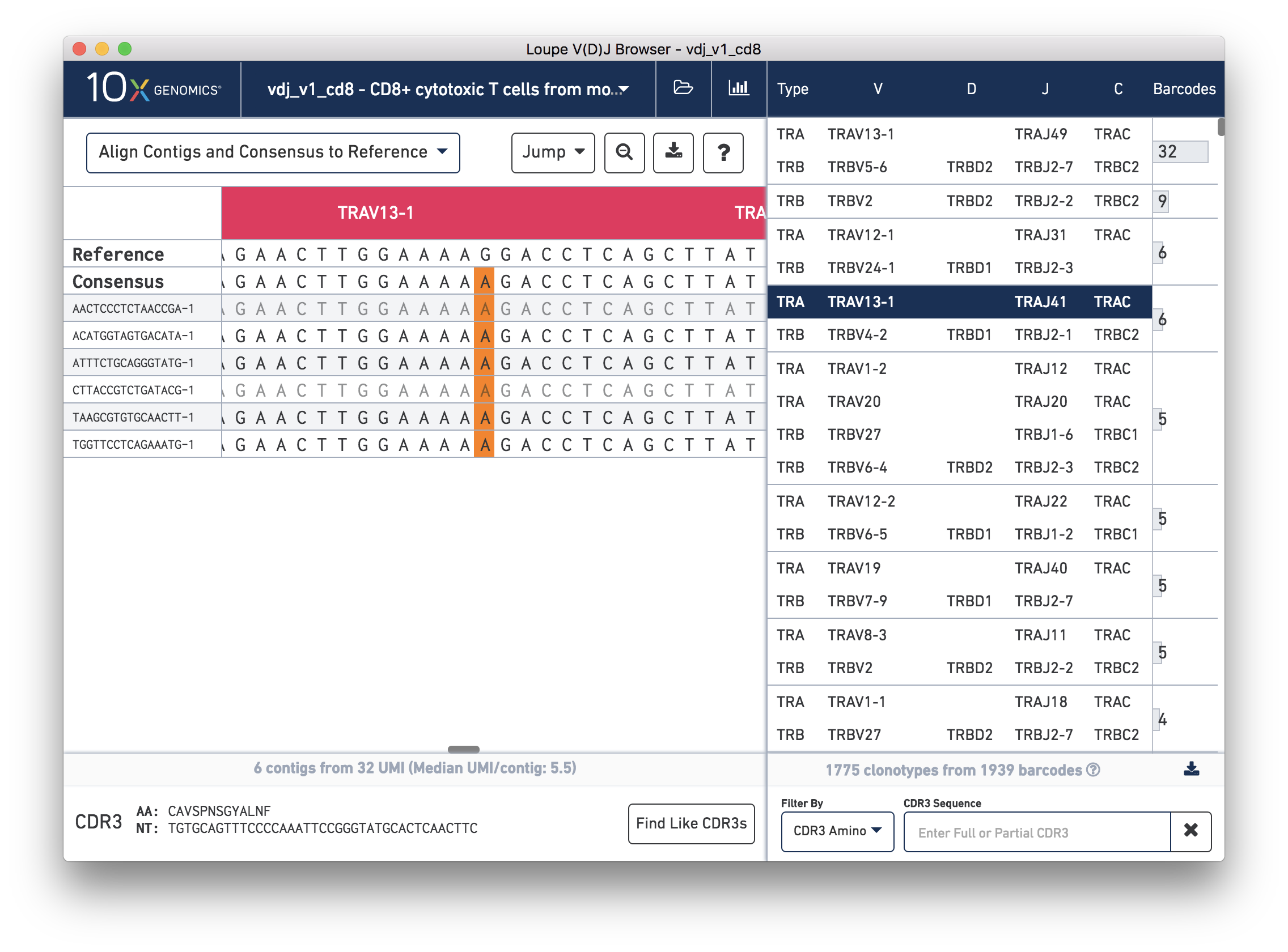
In this sequence level view, you can scroll side to side to view more of the sequence. Insertions still appear as blue vertical bars, but hovering over one will reveal the inserted sequence. The read quality is also indicated by the shade of the base, with darker letters indicating higher quality.
Since the sequences are long, it's easy to get lost scrolling side to side. The Jump button atop the gene annotations allows you to instantly navigate to the beginning of gene segments.
There are some additional helpful features of the single chain view we've not explored yet.
At the bottom of the Chain View, you can see the CDR3 for the chain in both amino acid and nucleotide sequences. The CDR3 shown is always the CDR3 of the consensus, regardless of the contig alignment setting. Clicking on either sequence will copy it to your clipboard for further documentation or investigation. Next to that you'll find the "Find Like CDR3s" button mentioned earlier, which filters the clonotype list to show all clonotypes which include a chain matching this CDR3.
In the top right of the Chain View panel you'll see a download button. Clicking on it will allow you to download all the sequences in this view (reference, consensus, and contigs) to a FASTA file as either nucleotides or amino acids. This allows for easy downstream analysis using standard bioinformatics tools.
Hovering over the barcode labels of any of the contigs will pop up a dialog with extra data about the contig including:
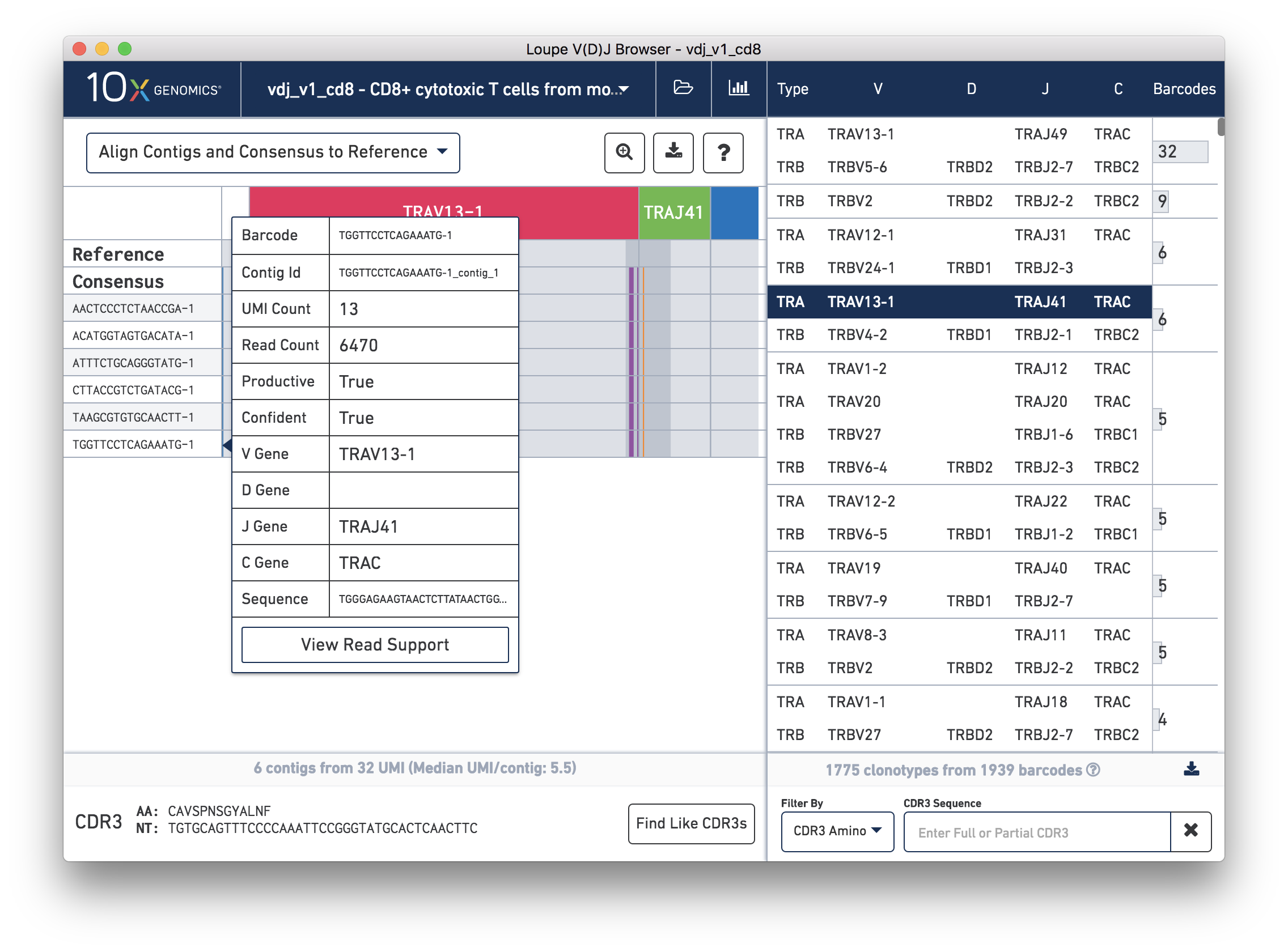
Clicking on any of these fields will copy them to your clipboard. It may be particularly useful to copy the contig sequence into the bioinformatics tools of your choice.
In this section, we've explored the consensus sequences and contigs that define a clonotype chain. Next, let's go one level farther, to explore how individual contigs were assembled.