Cell Ranger7.0, printed on 03/03/2025
This page describes the output file structure from the cellranger multi subcommand specifically for Fixed RNA Profiling (FRP) data.
Upon completion, the cellranger multi subcommand will produce an outs/ directory with the following structure:
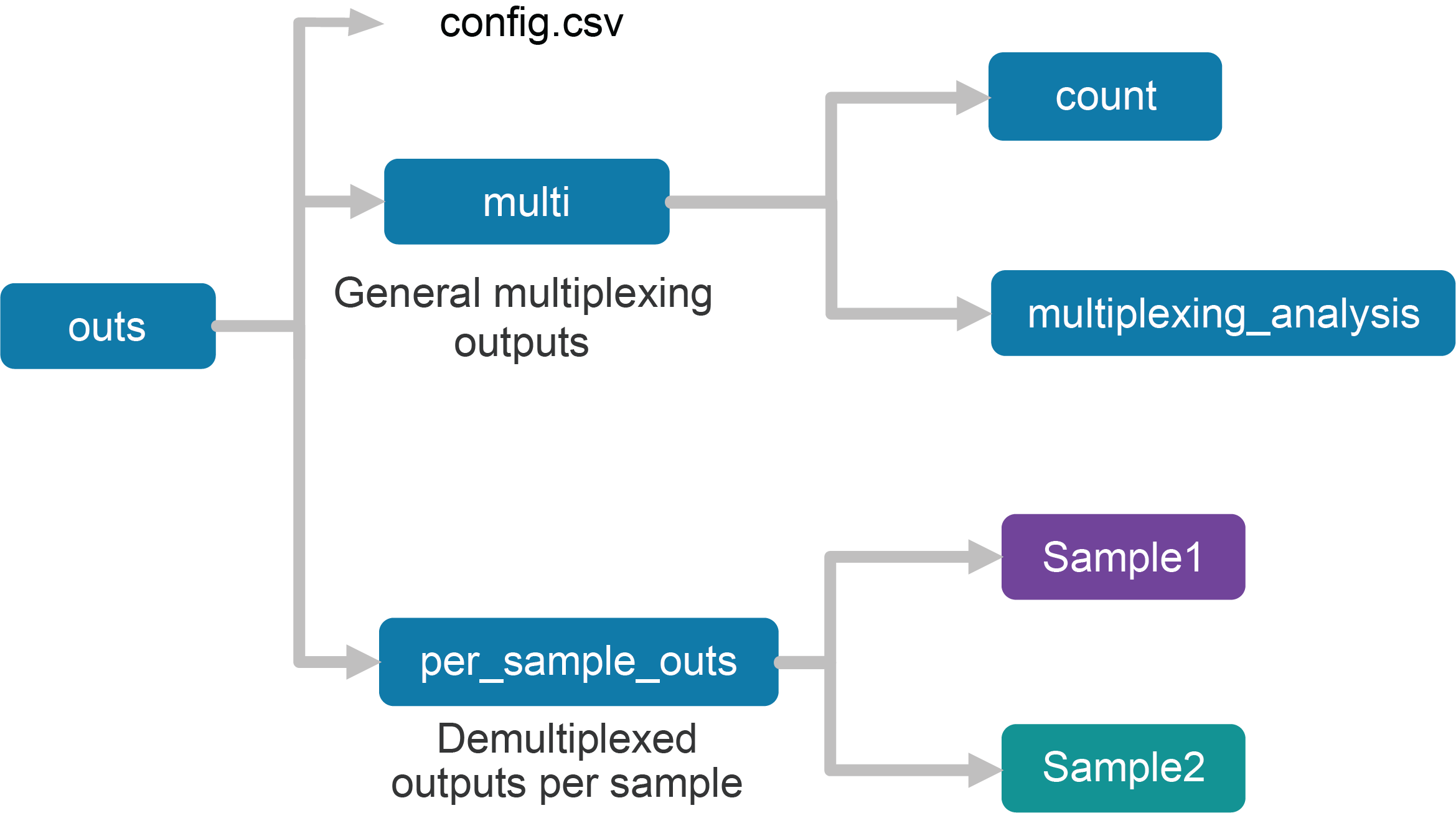
Using the tree Linux command, the file structure looks like this:
├── config.csv
├── multi
│ ├── count
│ └── multiplexing_analysis
└── per_sample_outs
├── Sample1
└── Sample2
The first section of the outputs contains the config.csv file, a duplicate of the input config CSV file. The files in the multi/ folder are generic to the entire FRP experiment, while the files in the per_sample_outs/ directory have been demultiplexed to single samples.
Within the multi/ directory, there are count and multiplexing_analysis directories. The multiplexing_analysis sub-directory is only generated in the context of multiplex Fixed RNA Profiling inputs.
└─ multi
├── count
│ ├── raw_cloupe.cloupe
│ ├── raw_feature_bc_matrix
│ │ ├── barcodes.tsv.gz
│ │ ├── features.tsv.gz
│ │ └── matrix.mtx.gz
│ ├── raw_feature_bc_matrix.h5
│ ├── raw_molecule_info.h5
│ ├── unassigned_alignments.bam
│ └── unassigned_alignments.bam.bai
└── multiplexing_analysis
├── cells_per_tag.json
└── frp_gem_barcode_overlap.csv
The count directory contains raw files that include cell-associated and background data from all samples within an experiment:
| Output File | Description |
|---|---|
raw_cloupe.cloupe |
A Loupe-readable file containing all cell-associated barcodes in the experiment. |
raw_feature_bc_matrix |
A matrix of UMI counts per (feature, barcode) pair, in MEX format. This matrix contains every barcode from the full fixed list of known good barcode sequences that has at least one read. This includes background and cell-associated barcodes from all samples as well as valid barcodes that were not assigned to a sample in case of a multiplex Fixed RNA Profiling experiment. Distinct ligation events are counted for Fixed RNA Profiling rather than distinct transcripts. |
raw_feature_bc_matrix.h5 |
Same information as raw_feature_bc_matrix in HDF5 format. |
raw_molecule_info.h5 |
Information about all molecules in the experiment. This file includes background and cell-associated barcodes from all samples as well as valid barcodes that were not assigned to a sample. This file cannot be used as input for cellranger aggr pipeline. |
unassigned_alignments.bam |
Reads with either with valid barcodes not assigned to a sample or invalid barcodes. |
unassigned_alignments.bam.bai |
Alignments from barcodes not assigned to any sample (index). |
The multiplexing_analysis directory contains:
| Output File | Description |
|---|---|
cells_per_tag.json |
Lists the cell-associated barcodes that were assigned a given Probe Barcode tag, for each tag, in JSON format. |
frp_gem_barcode_overlap.csv |
Contains the number of shared 10x GEM Barcodes for all pairs of observed Probe Barcode tags assigned to a sample. More details below. |
The cells_per_tag.json file looks like this, for each Probe Barcode (e.g., BC001) the cell-associated barcodes (e.g., "AACAAGCTCCCTCAAAACTTTAGG-1", etc.) are listed below it:
{
"BC001":[
"AACAAGCTCCCTCAAAACTTTAGG-1",
"AACATAGTCCCATAGCACTTTAGG-1",
"AACCAGGTCATGGTCCACTTTAGG-1",
...
"BC002":[
"AAACTGTCAGGAGCAAAACGGGAA-1",
"AAAGGGATCTAATCGTAACGGGAA-1",
"AACCAAATCGGTCAAGAACGGGAA-1",
...
}
The frp_gem_barcode_overlap.csv file can be used to troubleshoot scenarios where, for example, two different Probe Barcodes were accidentally added to the same hybridization reaction. An alert in the web summary will be triggered if the overlap coefficient is >60%.
Please contact [email protected] if you have questions.
barcode1_id,barcode2_id,barcode1_gems,barcode2_gems,common_gems,overlap BC001,BC002,271,311,1,0.003690037
Column descriptions:
barcode1_id: First Probe Barcode identifierbarcode2_id: Second Probe Barcode identifierbarcode1_gems: Number of 10x GEM Barcodes for barcode1barcode2_gems: Number of 10x GEM Barcodes for barcode2common_gems: Number of 10x GEM Barcodes in commonoverlap: The overlap coefficient of these two Probe Barcodes$$ \frac{\text{common_gems}}{\text{min(barcode1_gems, barcode2_gems)}} $$
The per_sample_outs directory contains sample-level files with any data associated with a valid Probe Barcode that could be assigned to a sample, including both cells and background.
├── count │ ├── analysis │ │ ├── clustering │ │ ├── diffexp │ │ ├── pca │ │ ├── tsne │ │ └── umap │ ├── probe_set.csv │ ├── sample_cloupe.cloupe │ ├── sample_alignments.bam │ ├── sample_alignments.bam.bai │ ├── sample_filtered_barcodes.csv │ ├── sample_filtered_feature_bc_matrix │ │ ├── barcodes.tsv.gz │ │ ├── features.tsv.gz │ │ └── matrix.mtx.gz │ ├── sample_filtered_feature_bc_matrix.h5 │ ├── sample_raw_feature_bc_matrix │ │ ├── barcodes.tsv.gz │ │ ├── features.tsv.gz │ │ └── matrix.mtx.gz │ ├── sample_raw_feature_bc_matrix.h5 │ └── sample_molecule_info.h5 ├── metrics_summary.csv └── web_summary.html
Note that the sample_filtered_feature_bc_matrix directory and sample_filtered_feature_bc_matrix.h5 file are similar to the filtered_feature_bc_matrix and filtered_feature_bc_matrix.h5, respectively, generated by |
The per_sample_outs directory contains:
| Output File | Description |
|---|---|
count/ |
Folder containing the results of any gene expression and Feature Barcode analysis, see table below. |
metrics_summary.csv |
Run summary metrics file in CSV format. Metric definitions available in the web summary ? help text. |
web_summary.html |
Run summary metrics and charts in HTML format, described in the multi web summary page. |
The count directory contains:
| Output File | Description |
|---|---|
analysis/ |
Folder containing the results of graph-based clusters and K-means clustering 2-10; differential gene expression analysis between clusters; and PCA, t-SNE, and UMAP dimensionality reduction. Learn more |
probe_set.csv |
A duplicate of the input probe_set.csv file. |
sample_cloupe.cloupe |
A Loupe Browser visualization and analysis file with cell-associated barcodes for the specific sample. |
sample_filtered_feature_bc_matrix/ |
Contains only detected cell-associated barcodes in MEX format. Each element of the matrix is the number of UMIs associated with a feature (row) and a barcode (column), as described in the feature-barcode matrix page. Distinct ligation events are counted for Fixed RNA Profiling rather than distinct transcripts. This file can be input into third-party packages and allows users to wrangle the barcode-feature matrix (e.g. to filter outlier cells, run dimensionality reduction, normalize gene expression). |
sample_filtered_feature_bc_matrix.h5 |
Same information as sample_filtered_feature_bc_matrix in HDF5 format. |
sample_molecule_info.h5 |
Contains per-molecule information for all molecules counted in the sample_raw_feature_bc_matrix for this sample that contain a valid barcode, valid UMI, and were assigned with high confidence to a gene or Feature Barcode. This file is a required input to run cellranger aggr. Learn more |
sample_alignments.bam |
Indexed BAM file containing position-sorted reads aligned to the genome and transcriptome, as well as unaligned reads, annotated with barcode information. Learn more |
sample_alignments.bam.bai |
Index file for the sample_alignments.bam |
sample_filtered_barcodes.csv |
File containing a list of only cell-associated barcodes. |
sample_raw_feature_bc_matrix/ |
Contains all barcodes assigned to this sample, including cell-associated and background barcodes in MEX format. Each element of the matrix is the number of UMIs associated with a feature (row) and a barcode (column), as described in the feature-barcode matrix page. Distinct ligation events are counted for Fixed RNA Profiling rather than distinct transcripts. This file is only generated in the context of multiplex Fixed RNA Profiling configuration. |
sample_raw_feature_bc_matrix_h5 |
Same information as sample_raw_feature_bc_matrix in HDF5 format. This file is only generated in the context of multiplex Fixed RNA Profiling configuration. |
Upon completion, the cellranger multi subcommand will produce an outs/ directory with the following structure:
├── config.csv
├── multi
│ └── count
└── per_sample_outs
└── Sample1
Within the multi directory, there is a count directory. This directory contains the same files as described above for the multiplex Fixed RNA Profiling count directory:
└─ multi
└── count
├── raw_cloupe.cloupe
├── raw_feature_bc_matrix
│ ├── barcodes.tsv.gz
│ ├── features.tsv.gz
│ └── matrix.mtx.gz
├── raw_feature_bc_matrix.h5
├── raw_molecule_info.h5
├── unassigned_alignments.bam
└── unassigned_alignments.bam.bai
The per_sample_outs directory contains data associated with all valid barcodes in the singleplex Fixed RNA Profiling library. This directory contains the same files as described above for the multiplex Fixed RNA Profiling per_sample_outs directory, except for the sample_raw_feature_bc_matrix/ and sample_raw_feature_bc_matrix.h5 files. The web_summary.html is described on the multi web summary page.
├── count │ ├── analysis │ │ ├── clustering │ │ ├── diffexp │ │ ├── pca │ │ ├── tsne │ │ └── umap │ ├── probe_set.csv │ ├── sample_cloupe.cloupe │ ├── sample_alignments.bam │ ├── sample_alignments.bam.bai │ ├── sample_filtered_barcodes.csv │ ├── sample_filtered_feature_bc_matrix │ │ ├── barcodes.tsv.gz │ │ ├── features.tsv.gz │ │ └── matrix.mtx.gz │ ├── sample_filtered_feature_bc_matrix.h5 │ └── sample_molecule_info.h5 ├── metrics_summary.csv └── web_summary.html
For Fixed RNA Profiling singleplex analyses with an Antibody Capture library, the per_sample_outs/count directory will also contain a feature_reference.csv, which is a duplicate of the input feature reference CSV file, and an antibody_analysis/ directory.
├── count │ ├── analysis │ │ ├── clustering │ │ ├── diffexp │ │ ├── pca │ │ ├── tsne │ │ └── umap │ ├── antibody_analysis/ │ │ └── aggregate_barcodes.csv │ ├── feature_reference.csv │ ├── ...
The web_summary.html is described on the multi web summary page.