Cell Ranger7.0, printed on 03/03/2025
Cell Ranger 6.0 and later supports analyzing 3' Cell Multiplexing data with the cellranger multi pipeline.
| New in Cell Ranger v7.0: Intronic reads are counted by default for whole transcriptome gene expression data. For more information, see our recommendation on including introns for gene expression analysis page. |
The cellranger multi pipeline is required to analyze 3' Cell Multiplexing data. Otherwise, users can continue to use cellranger count.
| 3' GEX | 3' FB | CellPlex | Use multi? |
|---|---|---|---|
| Yes | Yes | Yes | Required |
| Yes | Yes | No | Optional. Prefer count |
| Yes | No | Yes | Required |
| Yes | No | No | Optional. Prefer count |
| No | Yes | No | Optional. Prefer count |
| The Cell Ranger multi pipeline supports the analysis of cell multiplexed data (e.g., CellPlex). While custom tags are not supported by 10x Genomics, Cell Ranger is capable of analyzing cell multiplexed data using custom tags (such as TotalSeqA/B/C). Please see the CMO Reference section for more details. |
First, follow the instructions on running cellranger mkfastq to generate FASTQ files. For example, if the flow cell ID was HAWT7ADXX, then cellranger mkfastq will output FASTQ files in HAWT7ADXX/outs/fastq_path. If you are already starting with FASTQ files, you can skip this step and proceed directly to run cellranger multi.
Running cellranger multi requires a config CSV, described below, invoking the following arguments:
| Argument | Description |
|---|---|
--id | A unique run ID string: e.g. sample345 that is also the output folder name. Cannot be more than 64 characters. |
--csv | Path to config CSV file with input libraries and analysis parameters. |
The multi config CSV contains both the library definitions and experimental design variables. It is composed of up to four sections for 3' data:
Example formats for different product configurations are below. A template for a multi config CSV can be downloaded here and example multi config CSVs can be downloaded from 6.0 public datasets here.
Starting in Cell Ranger 7.0, the expected number of cells can either be auto-estimated or specified with expect-cells (e.g., to replicate a previous analysis), see Gene Expression algorithm overview. If needed, automated cell calling can be overridden with the force-cells option.
|
| Multi Config CSV | |
|---|---|
Section: [gene-expression] | |
| Field | Description |
reference
| Required. Path of folder containing 10x Genomics-compatible genome reference. |
min-assignment-confidence
| Optional. The minimum estimated likelihood to call a sample as tagged with a Cell Multiplexing Oligo (CMO) instead of "Unassigned". Users may wish to tolerate a higher rate of mis-assignment in order to obtain more singlets to include in their analysis, or a lower rate of mis-assignment at the cost of obtaining fewer singlets. By default, this value is 0.9. Contact [email protected] for further advice. |
cmo-set
| Optional. The default CMO reference IDs are built into the Cell Ranger software and do not need to be specified. However, this option can be used to specify the path to a custom CMO set CSV file, declaring CMO constructs and associated barcodes. See CMO Reference section for details. |
target-panel
| Optional. Path to a Target Panel CSV file or name of a 10x Genomics fixed gene panel (pathway, pan-cancer, immunology, neuroscience). Required for Targeted Gene Expression analysis. Default analysis will exclude intronic mapped reads, which is the recommended mode for targeted assays. |
no-target-umi-filter
| Optional. Disable targeted UMI filtering stage. See Targeted Algorithms for details. Default: false |
r1-length
| Optional. Limit the length of the input Read 1 sequence of Gene Expression libraries to the first N bases, where N is a user-supplied value. Note that the length includes the 10x Barcode and UMI sequences so do not set this below 26. This and r2-length are useful options for determining the optimal read length for sequencing. Default: do not trim Read 1. |
r2-length
| Optional. Limit the length of the input Read 2 sequence of Gene Expression libraries to the first N bases, where N is a user-supplied value. Trimming occurs before sequencing metrics are computed and therefore, limiting the length of Read 2 may affect Q30 scores. Default: do not trim Read 2. |
chemistry
| Optional. Assay configuration. NOTE: by default, the assay configuration is detected automatically (recommended mode). Users will typically not need to specify a chemistry. Options are: auto for autodetection
threeprime for Single Cell 3'
SC3Pv1 or SC3Pv2 or SC3Pv3 for Single Cell 3' v1/v2/v3
SC3Pv3HT for Single Cell 3' v3.1 HT
SC-FB for Single Cell Antibody-only 3' v2
Default: auto |
expect-cells
| Optional. Override the pipeline’s auto-estimate. See cell calling algorithm overview for details on how this parameter is used. If used, enter the expected number of recovered cells. Up to 30,000 cells are supported with standard kits for Cell Multiplexing and up to 60,000 cells for HT kits. |
force-cells
| Optional. Force pipeline to use this number of cells, bypassing cell detection. Default: detect cells using Cell Ranger's cell calling algorithm |
include-introns
| Optional. Set to false to exclude intronic reads in count. Including introns in analysis is recommended to maximize sensitivity, except when target-panel is used. Default: true |
no-secondary
| Optional. Disable secondary analysis, e.g. clustering. Default: false |
no-bam
| Optional. Set this flag to true to skip BAM file generation. This will reduce the total computation time for the pipestance and the size of the output directory. If unsure, we recommend not using this option, as BAM files can be useful for troubleshooting and downstream analysis. Default: false |
check-library-compatibility
| Optional. This option allows users to disable the check that evaluates 10x Barcode overlap between libraries when multiple libraries are specified (e.g., Gene Expression + Antibody Capture). Setting this option to false will disable the check across all library combinations. We recommend running this check (default), however if the pipeline errors out, users can bypass the check to generate outputs for troubleshooting. Default: true |
barcode-sample-assignment
| Optional. Path to a barcode-sample assignment CSV file that specifies the barcodes that belong to each sample. See details below to set up this file. |
Section: [feature] | |
| Field | Description |
reference
| Path to the Feature reference CSV file, declaring Feature Barcode constructs and associated barcodes. Required only for Antibody Capture or CRISPR Guide Capture libraries. |
r1-length
| Optional. Limit the length of the input Read 1 sequence of Feature Barcode libraries (Antibody Capture, CRISPR Guide Capture, Multiplexing Capture) to the first N bases, where N is a user-supplied value. Note that the length includes the 10x Barcode and UMI sequences so do not set this below 26. This and r2-length are useful options for determining the optimal read length for sequencing. Default: do not trim Read 1. |
r2-length
| Optional. Limit the length of the input Read 2 sequence of Feature Barcode libraries (Antibody Capture, CRISPR Guide Capture, Multiplexing Capture) to the first N bases, where N is a user-supplied value. Trimming occurs before sequencing metrics are computed and therefore, limiting the length of Read 2 may affect Q30 scores. Default: do not trim Read 2. |
Section: [libraries](see also Specifying Input FASTQ Files for cellranger multi) | |
| Column | Description |
fastq_id
| Required. The Illumina sample name to analyze. This will be as specified in the sample sheet supplied to mkfastq or bcl2fastq. |
fastqs
| Required. Path to the folder containing the FASTQ files to be analyzed. Generally, this will be the fastq_path folder generated by cellranger mkfastq. |
feature_types
| Required. The underlying feature type of the library, which must be one of:
Gene Expression
Antibody Capture
CRISPR Guide Capture
Multiplexing Capture |
lanes
| Optional. The lanes associated with this sample, separated with a pipe (e.g., 1|2). Default: uses all lanes |
physical_library_id
| Optional. Library type. NOTE: by default, the library type is detected automatically based on specified feature_types (recommended mode). Users typically do not need to include the physical_library_id column in the CSV file. |
subsample_rate
| Optional. The rate at which reads from the provided FASTQ files are sampled. Must be strictly greater than 0 and less than or equal to 1. |
Section: [samples] | |
| Column | Description |
sample_id
| A name to identify a multiplexed sample. Must be alphanumeric with hyphens and/or underscores, and less than 64 characters. Required for Cell Multiplexing libraries. |
cmo_ids
| The Cell Multiplexing oligo IDs used to multiplex this sample. If multiple CMOs were used for a sample, separate IDs with a pipe (e.g., CMO301|CMO302). Required for Cell Multiplexing libraries. |
description
| Optional. A description for the sample. |
For help on how to configure the [libraries] section to target a particular set of FASTQs, consult Specifying Input FASTQ Files for cellranger multi.
|
cd /home/jdoe/runs cellranger multi --id=sample345 --csv=/home/jdoe/sample345.csv
Martian Runtime - v4.0.8 Running preflight checks (please wait)... ...
By default, Cell Ranger will use all of the cores available on your
system to execute pipeline stages. You can specify a different number of cores
to use with the --localcores option; for example, --localcores=16
will limit Cell Ranger to using up to sixteen cores at once. Similarly,
--localmem will restrict the amount of memory (in GB) used by
Cell Ranger.
The pipeline will create a new folder named with the run ID you specified using the --id argument (e.g. /home/jdoe/runs/sample345) for its output. If this folder already exists, Cell Ranger will assume it is an existing pipestance and attempt to resume running it.
Waiting 6 seconds for UI to do final refresh. Pipestance completed successfully! yyyy-mm-dd hh:mm:ss Shutting down. Saving pipestance info to "tiny/tiny.mri.tgz"
sample345). The subfolder named outs/ will contain the main pipeline outputs.Here are a few example multi config CSVs for some common product configurations, along with simplified diagrams for the corresponding experimental set up. Make sure to replace /path/to with the actual full path to your data, and edit any text in red according to the experiment's sample/library/file names.
| Experimental Design | Multi config CSV |
3' GEX with Cell Multiplexing, 1 CMO/sample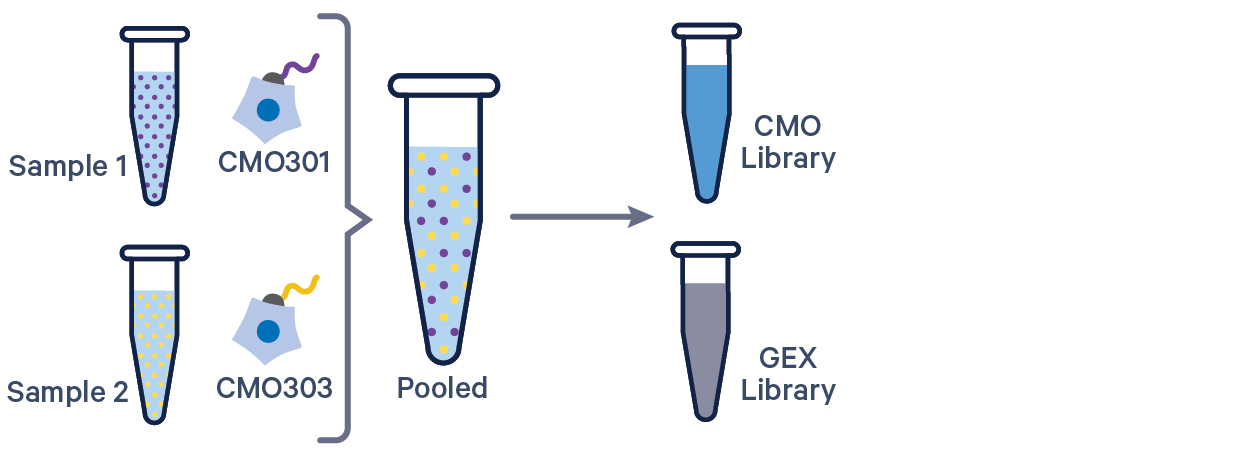 See example dataset |
[gene-expression] reference,/path/to/transcriptome |
3' GEX with Cell Multiplexing, multiple CMOs/sample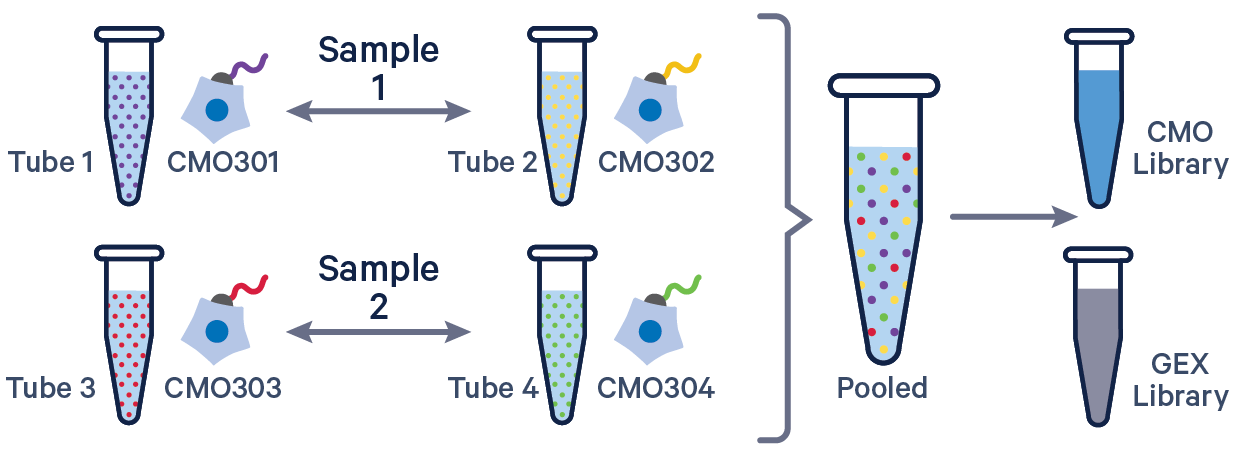 See example dataset. Note usage of the | to separate CMO tags. Learn more about when to use multiple CMOs per sample here. |
[gene-expression] reference,/path/to/transcriptome |
3' GEX with Cell Multiplexing and Feature Barcode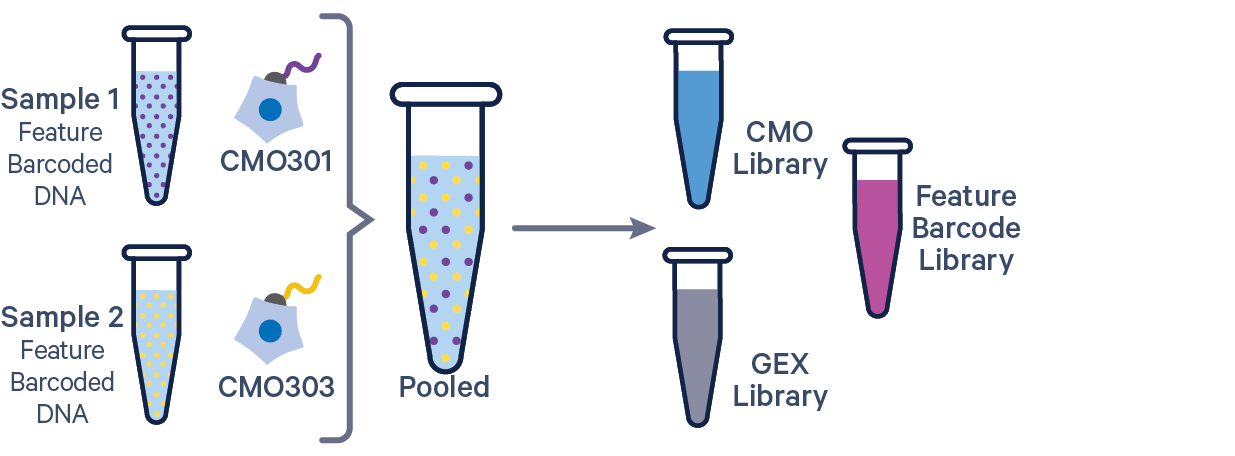 The Feature Barcode in this config CSV example ( [libraries] section) is Antibody Capture. Use CRISPR Guide Capture for CRISPR Feature Barcode experiments. |
[gene-expression] reference,/path/to/transcriptome |
The cmo-set option in the [gene-expression] section of the multi config CSV allows you to provide a reference for custom Cell Multiplexing oligos (e.g., antibody TotalSeqA/B/C tags). The design of this reference is nearly identical to the Feature Barcode Reference used to describe Feature Barcodes, with one difference: the feature_type is required to be Multiplexing Capture instead of those feature types supported in the Feature Barcode reference. The id column may contain alphanumeric, underscore, and hyphen characters; special characters like a pipe (|) should not be used in this file (only for separating multiple CMO IDs from the same sample in config CSV).
For example, Cell Ranger's default CMO reference looks like this (built into Cell Ranger):
id,name,read,pattern,sequence,feature_type CMO301,CMO301,R2,5P(BC),ATGAGGAATTCCTGC,Multiplexing Capture CMO302,CMO302,R2,5P(BC),CATGCCAATAGAGCG,Multiplexing Capture CMO303,CMO303,R2,5P(BC),CCGTCGTCCAAGCAT,Multiplexing Capture CMO304,CMO304,R2,5P(BC),AACGTTAATCACTCA,Multiplexing Capture CMO305,CMO305,R2,5P(BC),CGCGATATGGTCGGA,Multiplexing Capture CMO306,CMO306,R2,5P(BC),AAGATGAGGTCTGTG,Multiplexing Capture CMO307,CMO307,R2,5P(BC),AAGCTCGTTGGAAGA,Multiplexing Capture CMO308,CMO308,R2,5P(BC),CGGATTCCACATCAT,Multiplexing Capture CMO309,CMO309,R2,5P(BC),GTTGATCTATAACAG,Multiplexing Capture CMO310,CMO310,R2,5P(BC),GCAGGAGGTATCAAT,Multiplexing Capture CMO311,CMO311,R2,5P(BC),GAATCGTGATTCTTC,Multiplexing Capture CMO312,CMO312,R2,5P(BC),ACATGGTCAACGCTG,Multiplexing Capture
The default CMO reference above is available as a downloadable CSV here.
The barcode-sample-assignment option in the [gene-expression] section of the multi config CSV allows users to provide a file that manually specifies the barcodes for each sample. It will override Cell Ranger's default cell calling and tag calling steps, and may be useful in cases where data with microfluidic failures can be partially rescued. This feature allows users to import custom tag calling done via 3rd party tools as well (see the Tag assignment of 10x Genomics CellPlex data using Seurat's HTODemux function Analysis Guide for help).
Here is an example multi config CSV:
[gene-expression] reference,/path/to/transcriptome barcode-sample-assignment,/path/to/barcode_sample_assignment.csv
[libraries] fastq_id,fastqs,feature_types gex1,/path/to/fastqs,Gene Expression mux1,/path/to/fastqs,Multiplexing Capture
[samples] sample_id,cmo_ids sample1,CMO301 sample2,CMO303
The barcode-sample CSV file has at most two columns, one for the barcode sequence and another that is either the sample ID or the tag assignment. A barcode can only be assigned to one sample; barcodes with multiple sample or tag entries will result in an error in Cell Ranger. Here are two examples:
Barcode,Sample_ID ACGTACGTACGTACGT-1,Jurkat CGTACGTACGTACGTA-1,Raji GTACGTACGTACGTAC-1,Jurkat TACGTACGTACGTACG-1,Raji ...
Barcode,Assignment ACGTACGTACGTACGT-1,CMO1 CGTACGTACGTACGTA-1,Multiplet GTACGTACGTACGTAC-1,Blank TACGTACGTACGTACG-1,Unassigned ...