Cell Ranger7.0, printed on 04/10/2025
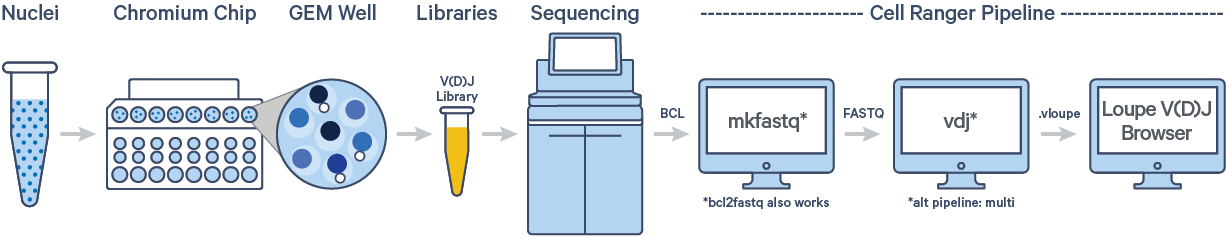
The cellranger vdj pipeline can be used to analyze sequencing data produced from Chromium Single Cell 5′ V(D)J libraries. It takes FASTQ files from cellranger mkfastq, BCL Convert or bcl2fastq for V(D)J libraries and performs sequence assembly and paired clonotype calling. It uses the Chromium cellular barcodes and UMIs to assemble V(D)J transcripts per cell. Clonotypes and CDR3 sequences are output as a .vloupe file which can be loaded into Loupe V(D)J Browser.
| Do you have single cell gene expression (GEX) libraries and V(D)J libraries from the same sample? Check out the cellranger multi pipeline page to analyze both libraries together. cellranger multi is the recommended pipeline to analyze V(D)J+GEX data from the same sample. |
To generate FASTQ files, refer to the instructions on running cellranger mkfastq. For help getting started, try the cellranger vdj tutorial.
For a complete list of cellranger vdj command-line arguments, run cellranger vdj --help.
To generate single cell V(D)J sequences and annotations for a single library, run cellranger vdj with these required arguments:
| For help on which arguments to use to target a particular set of FASTQs, consult Specifying Input FASTQ Files for cellranger count and vdj. |
| Argument | Description |
|---|---|
--id | Required. A unique run ID string: e.g. sample345 |
--fastqs | Required. Path of the FASTQ folder generated by cellranger mkfastq e.g. /home/jdoe/runs/HAWT7ADXX/outs/fastq_path
Can take multiple comma-separated paths, which is helpful if the same library was sequenced on multiple flowcells. Doing this will treat all reads from the library, across flowcells, as one sample. |
--reference | Required. Path to the Cell Ranger V(D)J compatible reference e.g. /opt/refdata-cellranger-vdj-GRCh38-alts-ensembl-7.0.0. If --denovo is specified, this parameter is optional. |
--sample | Required. Sample name as specified in the sample sheet supplied to mkfastq.
Can take multiple comma-separated values, which is helpful if the sample was sequenced on multiple flowcells and the sample name used (and therefore fastq file prefix) is not identical between them. Doing this will treat all reads from the library, across flowcells, as one sample. |
| Option | Description |
|---|---|
--denovo | Optional. If specified, this flag prevents the use of V(D)J reference during the assembly process. --reference is optional. If --denovo is specified and --reference is not, the --inner_enrichment_primers argument is required. The --denovo option is most useful for full de novo assembly without a V(D)J reference. If you have a V(D)J reference, using --denovo will yield similar but slightly degraded results. |
--inner-enrichment-primers | Optional. This flag takes a .txt file containing primer sequences that were used to enrich cDNA for V(D)J sequences. The primers must be listed one per line. If two sets of primers were used for amplification, the .txt file must contain only the innermost reverse PCR primers that are complementary to the constant region. An example .txt file for human TCR dataset would have these lines:
AGTCTCTCAGCTGGTACACG TCTGATGGCTCAAACACAGC The |
| Option | Description |
|---|---|
--localcores | Optional. Restricts cellranger to use the specified number of cores to execute pipeline stages. By default, cellranger will use all of the cores available on your system. |
--localmem | Optional. Restricts cellranger to use the specified amount of memory (in GB) to execute pipeline stages. By default, cellranger will use 90% of the memory available on your system. |
| Option | Description |
|---|---|
--lanes | Optional. Lanes associated with this sample |
| Option | Description |
|---|---|
--chain |
Optional. Force the analysis to be carried out for a particular chain type. The accepted values are:
|
After determining your input arguments and options, run cellranger vdj:
$ cd /home/jdoe/runs $ cellranger vdj --id=sample345 \ --reference=/opt/refdata-cellranger-vdj-GRCh38-alts-ensembl-7.0.0 \ --fastqs=/home/jdoe/runs/HAWT7ADXX/outs/fastq_path \ --sample=mysample \ --localcores=8 \ --localmem=64
Following a set of preflight checks to validate input arguments, cellranger vdj pipeline stages will begin to run:
Martian Runtime - v4.0.8 Running preflight checks (please wait)... yyyy-mm-dd hh:mm:ss [runtime] (ready) ID.sample345.SC_VDJ_ASSEMBLER_CS.VDJ_PREFLIGHT yyyy-mm-dd hh:mm:ss [runtime] (run:local) ID.sample345.SC_VDJ_ASSEMBLER_CS.VDJ_PREFLIGHT.fork0.chnk0.main yyyy-mm-dd hh:mm:ss [runtime] (ready) ID.sample345.SC_VDJ_ASSEMBLER_CS.VDJ_PREFLIGHT_LOCAL ...
By default, cellranger will use all of the cores available on your
system to execute pipeline stages. You can specify a different number of cores
to use with the --localcores option; for example, --localcores=16
will limit cellranger to using up to sixteen cores at once. Similarly,
--localmem will restrict the amount of memory (in GB) used by
cellranger.
The pipeline will create a new folder named with the sample ID you specified (e.g. /home/jdoe/runs/sample345) for its output. If this folder already exists, cellranger will assume it is an existing pipestance and attempt to resume running it.
A successful cellranger vdj run should conclude with a message similar to this:
Outputs:
- Run summary HTML: /home/jdoe/runs/sample345/outs/web_summary.html
- Run summary CSV: /home/jdoe/runs/sample345/outs/metrics_summary.csv
- Clonotype info: /home/jdoe/runs/sample345/outs/clonotypes.csv
- Filtered contig sequences FASTA: /home/jdoe/runs/sample345/outs/filtered_contig.fasta
- Filtered contig sequences FASTQ: /home/jdoe/runs/sample345/outs/filtered_contig.fastq
- Filtered contigs (CSV): /home/jdoe/runs/sample345/outs/filtered_contig_annotations.csv
- All-contig FASTA: /home/jdoe/runs/sample345/outs/all_contig.fasta
- All-contig FASTA index: /home/jdoe/runs/sample345/outs/all_contig.fasta.fai
- All-contig FASTQ: /home/jdoe/runs/sample345/outs/all_contig.fastq
- Read-contig alignments: /home/jdoe/runs/sample345/outs/all_contig.bam
- Read-contig alignment index: /home/jdoe/runs/sample345/outs/all_contig.bam.bai
- All contig annotations (JSON): /home/jdoe/runs/sample345/outs/all_contig_annotations.json
- All contig annotations (BED): /home/jdoe/runs/sample345/outs/all_contig_annotations.bed
- All contig annotations (CSV): /home/jdoe/runs/sample345/outs/all_contig_annotations.csv
- Barcodes that are declared to be targetted cells: /home/jdoe/runs/sample345/outs/cell_barcodes.json
- Clonotype consensus FASTA: /home/jdoe/runs/sample345/outs/consensus.fasta
- Clonotype consensus FASTA index: /home/jdoe/runs/sample345/outs/consensus.fasta.fai
- Contig-consensus alignments: /home/jdoe/runs/sample345/outs/consensus.bam
- Contig-consensus alignment index: /home/jdoe/runs/sample345/outs/consensus.bam.bai
- Clonotype consensus annotations (CSV): /home/jdoe/runs/sample345/outs/consensus_annotations.csv
- Concatenated reference sequences: /home/jdoe/runs/sample345/outs/concat_ref.fasta
- Concatenated reference index: /home/jdoe/runs/sample345/outs/concat_ref.fasta.fai
- Contig-reference alignments: /home/jdoe/runs/sample345/outs/concat_ref.bam
- Contig-reference alignment index: /home/jdoe/runs/sample345/outs/concat_ref.bam.bai
- Loupe V(D)J Browser file: /home/jdoe/runs/sample345/outs/vloupe.vloupe
- V(D)J reference:
fasta:
regions: /home/jdoe/runs/sample345/outs/vdj_reference/fasta/regions.fa
donor_regions: /home/jdoe/runs/sample345/outs/vdj_reference/fasta/donor_regions.fa
reference: /home/jdoe/runs/sample345/outs/vdj_reference/reference.json
- AIRR Rearrangement TSV: /home/jdoe/runs/sample345/outs/airr_rearrangement.tsv
- All contig info (ProtoBuf format): /home/jdoe/runs/sample345/outs/vdj_contig_info.pb
Waiting 6 seconds for UI to do final refresh.
Pipestance completed successfully!
The output folder name is the same as the sample ID you specified (e.g. sample345). The outs subfolder contains the main pipeline output files.
Once cellranger vdj has successfully completed, you can browse the resulting summary HTML file in any supported web browser,
open the .vloupe file in Loupe V(D)J Browser, or refer to the Understanding Output section to explore the data by hand.