Cell Ranger7.1, printed on 04/11/2025
In this tutorial, you will:
To follow along, you must:
The Chromium Single Cell 5’ Barcode Enabled Antigen Mapping (BEAM) technology offers a scalable approach for mapping a V(D)J receptor to a target antigen by enabling the detection of gene expression profiles, paired V(D)J receptors, and signal from a bound antigen from the same single cell. All of these libraries, generated from a single GEM well, can be analyzed together with Cell Ranger v7.1 or later using the cellranger multi pipeline.
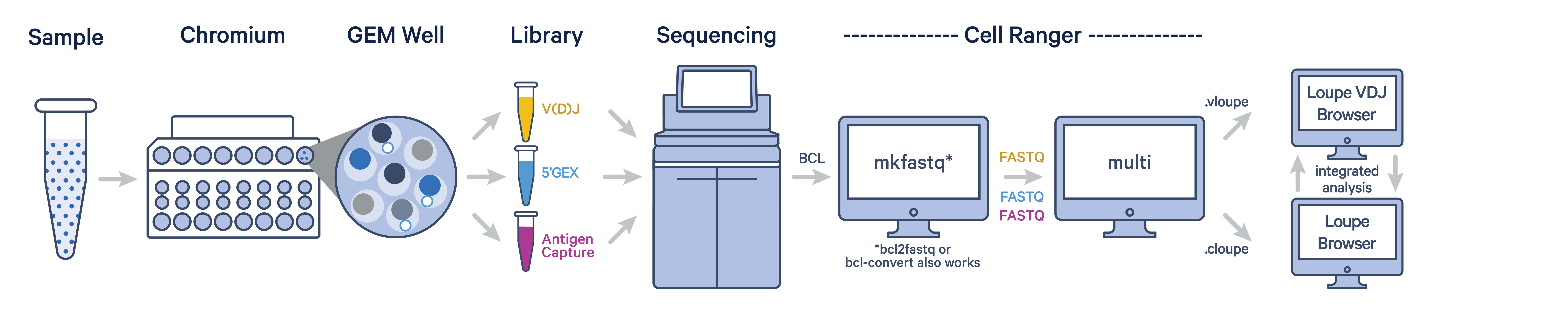
We will work with the 2k Transgenic HEL Mouse Splenocytes (BEAM-Ab) dataset.
Open up a terminal window. You may log in to a remote server or choose to perform the compute on your local machine. Refer to the System Requirements page for details.
In the working directory, create a new folder called beam-ab and cd into that folder:
mkdir beam-ab cd beam-ab
Download the input FASTQ files:
The FASTQs come as a .tar compression (19.8 GB) and may take over ten minutes to download.
curl -O https://cf.10xgenomics.com/samples/cell-vdj/7.1.0/2k_BEAM-Ab_Mouse_HEL_5pv2_Multiplex/2k_BEAM-Ab_Mouse_HEL_5pv2_Multiplex_fastqs.tar
A file named 2k_BEAM-Ab_Mouse_HEL_5pv2_Multiplex_fastqs.tar should appear in your directory when you list files with the ls -lt command.
Uncompress the FASTQs:
tar -xvf 2k_BEAM-Ab_Mouse_HEL_5pv2_Multiplex_fastqs.tar
You should now see a folder called 2k_BEAM-Ab_Mouse_HEL_5pv2_fastqs:
cd 2k_BEAM-Ab_Mouse_HEL_5pv2_fastqs ls
The folder contains three subfolders with library-specific FASTQS files: antigen_capture, gex, and vdj.
Navigate back to the working directory:
cd ..
Double check you are in the correct directory by running the ls command; the working directory should have the FASTQs 2k_BEAM-Ab_Mouse_HEL_5pv2_fastqs folder.
Download the Feature Reference CSV available for this example dataset.
curl -O https://cf.10xgenomics.com/samples/cell-vdj/7.1.0/2k_BEAM-Ab_Mouse_HEL_5pv2_Multiplex/2k_BEAM-Ab_Mouse_HEL_5pv2_Multiplex_count_feature_reference.csv
To view the contents of the Feature Reference CSV, open it in your text editor of choice (e.g., nano)
nano 2k_BEAM-Ab_Mouse_HEL_5pv2_Multiplex_count_feature_reference.csv
The contents should look like this:
id,name,read,pattern,sequence,feature_type SARS-TRI-S_WT,SARS-TRI-S_WT,R2,^(BC),CGATGCCGGACGATC,Antigen Capture Anti-Hen_Egg_Lysozyme,Anti-Hen_Egg_Lysozyme,R2,^(BC),CCGTCTCACCGATAT,Antigen Capture gp120,gp120,R2,^(BC),GATTGGCTACTCAAT,Antigen Capture H5N1,H5N1,R2,^(BC),CGGCTCACCGCGTCT,Antigen Capture negative_control,negative_control,R2,^(BC),CTATCTACCGGCTCG,Antigen Capture
Since this is a BEAM-Ab (BCR Antigen Capture) dataset, the Feature Reference CSV does NOT contain the additional mhc_allele column. The BEAM-T tutorial guides you through analyzing a TCR Antigen Capture dataset.
You do not need to change the Feature Reference CSV for this tutorial. Remember to customize it when working with your own data. Learn more about the Feature Reference CSV.
Download the pre-built mouse reference transcriptome in the working directory (beam-ab/) and uncompress it:
curl -O https://cf.10xgenomics.com/supp/cell-vdj/refdata-gex-mm10-2020-A.tar.gz tar -xvf refdata-gex-mm10-2020-A.tar.gz
A custom-made mouse V(D)J reference was used as input. This reference differs from the pre-built mouse reference in the sequence of only one V(D)J gene. For more information, please contact support@10xgenomics.com.
Download the custom built mouse V(D)J reference in the working directory and uncompress it:
curl -O https://cf.10xgenomics.com/samples/cell-vdj/7.1.0/2k_BEAM-Ab_Mouse_HEL_5pv2_Multiplex/2k_BEAM-Ab_Mouse_HEL_5pv2_Multiplex_vdj_reference.tar.gz tar -xvf 2k_BEAM-Ab_Mouse_HEL_5pv2_Multiplex_vdj_reference.tar.gz
In your working directory, create a new CSV file called 2k_BEAM-Ab_Mouse_HEL_5pv2_Multiplex_config.csv using your text editor of choice. For example, you can create a file with nano using this command:
nano 2k_BEAM-Ab_Mouse_HEL_5pv2_Multiplex_config.csv
Copy and paste this text into the newly created file and customize the /path/to/... part of file paths:
[gene-expression] ref,/path/to/references/refdata-gex-mm10-2020-A [feature] ref,/path/to/feature_references/2k_BEAM-Ab_Mouse_HEL_5pv2_Multiplex_count_feature_reference.csv [vdj] ref,/path/to/references/vdj_reference [libraries] fastq_id,fastqs,lanes,feature_types beamab_mouse_hel_ag,/path/to/fastqs/2k_BEAM-Ab_Mouse_HEL_5pv2_fastqs/antigen_capture,1|2|3|4,Antigen Capture beamab_mouse_hel_vdj,/path/to/fastqs/2k_BEAM-Ab_Mouse_HEL_5pv2_fastqs/vdj,1|2|3|4,VDJ-B beamab_mouse_hel_gex,/path/to/fastqs/2k_BEAM-Ab_Mouse_HEL_5pv2_fastqs/gex,1|2|3|4,Gene Expression [antigen-specificity] control_id negative_control
Use your text editor's save command to save the file. In nano, save by typing → → .
A customizable multi config CSV template is available for download on the example dataset page, under the Input Files tab.
Once you have all the necessary files, make a new directory called runs/ in your beam-ab/ working directory:
mkdir runs/ cd runs/
You will run cellranger multi in the runs/ directory.
After downloading/creating the FASTQ files, Feature Reference CSV, reference transcriptome, and V(D)J reference, you are ready to run cellranger multi.
Print the usage statement to get a list of all the options:
cellranger multi --help
The output should look similar to:
user_prompt$ cellranger multi --help
cellranger-multi
Analyze multiplexed data or combined gene expression/immune profiling/feature
barcode data
USAGE:
cellranger multi [FLAGS] [OPTIONS] --id --csv
FLAGS:
--dry Do not execute the pipeline. Generate a pipeline
invocation (.mro) file and stop
--disable-ui Do not serve the web UI
--noexit Keep web UI running after pipestance completes or fails
--nopreflight Skip preflight checks
-h, --help Prints help information
OPTIONS:
--id A unique run id and output folder name [a-zA-Z0-
9_-]+
--description Sample description to embed in output files
[default: ]
--csv Path of CSV file enumerating input libraries and
analysis parameters
--jobmode Job manager to use. Valid options: local
(default), sge, lsf, slurm or path to a
.template file. Search for help on "Cluster
Mode" at support.10xgenomics.com for more
details on configuring the pipeline to use a
compute cluster [default: local]
--localcores Set max cores the pipeline may request at one
time. Only applies to local jobs
....
| Option | Description |
|---|---|
--id |
The id argument must be a unique run ID. We will call this run HumanB_Cell_multi based on the sample type in the example dataset. |
--csv |
Path to the multi config CSV file enumerating input libraries and analysis parameters. Your multi_config.csv file is in the working directory. When executing cellranger multi from the runs directory, the relative path should be: ../multi_config.csv |
From within the beam-ab/runs/ directory, run cellranger multi
/path/to/cellranger-7.1.0/cellranger multi --id=beam-ab-run --csv=../2k_BEAM-Ab_Mouse_HEL_5pv2_Multiplex_config.csv
The run begins similarly to this:
Martian Runtime - v4.0.10 2023-06-15 11:44:24 [jobmngr] WARNING: configured to use 334GB of local memory, but only 194.9GB is currently available. Serving UI at http://bespin3.fuzzplex.com:34513?auth=-Sm5gsg6_G8FjcUX0_YD5J8SYoBODz4IWoVIK9ec0jg Running preflight checks (please wait)... 2023-06-15 11:44:33 [runtime] (ready) ID.beam-ab-run.SC_MULTI_CS.PARSE_MULTI_CONFIG 2023-06-15 11:44:33 [runtime] (run:local) ID.beam-ab-run.SC_MULTI_CS.PARSE_MULTI_CONFIG.fork0.chnk0.main 2023-06-15 11:44:56 [runtime] (chunks_complete) ID.beam-ab-run.SC_MULTI_CS.PARSE_MULTI_CONFIG 2023-06-15 11:44:56 [runtime] (ready) ID.beam-ab-run.SC_MULTI_CS.FULL_COUNT_INPUTS.WRITE_GENE_INDEX 2023-06-15 11:44:56 [runtime] (run:local) ID.beam-ab-run.SC_MULTI_CS.FULL_COUNT_INPUTS.WRITE_GENE_INDEX.fork0.chnk0.main ....
When the output of the cellranger multi command says, “Pipestance completed successfully!”, the job is done:
web_summary: /jane.doe/beam-ab/runs/beam-ab-run/outs/per_sample_outs/beam-ab/web_summary.html
metrics_summary: /jane.doe/beam-ab/runs/beam-ab-run/runs/beam-ab-run/outs/per_sample_outs/beam-ab/metrics_summary$
}
Waiting 6 seconds for UI to do final refresh.
Pipestance completed successfully!
A successful cellranger multi run produces a new directory called beam-ab-run (based on the --id flag specified during the run). The contents of the beam-ab-run directory:
. ├── beam-ab-run │ ├── beam-ab.mri.tgz │ ├── _cmdline │ ├── _filelist │ ├── _finalstate │ ├── _invocation │ ├── _jobmode │ ├── _log │ ├── _mrosource │ ├── outs │ ├── _perf │ ├── _perf._truncated_ │ ├── SC_MULTI_CS │ ├── _sitecheck │ ├── _tags │ ├── _timestamp │ ├── _uuid │ ├── _vdrkill │ └── _versions
The outs/ directory contains all important output files generated by the cellranger multi pipeline:
── runs
└── beam-ab-run
└──outs
├── config.csv
├── multi
│ ├── count
│ │ ├── feature_reference.csv
│ │ ├── raw_cloupe.cloupe
│ ├── raw_feature_bc_matrix
│ │ ├── raw_feature_bc_matrix.h5
│ │ ├── raw_molecule_info.h5
│ │ ├── unassigned_alignments.bam
│ │ └── unassigned_alignments.bam.bai
│ └── vdj_b
│ ├── all_contig_annotations.bed
│ ├── all_contig_annotations.csv
│ ├── all_contig_annotations.json
│ ├── all_contig.bam
│ ├── all_contig.bam.bai
│ ├── all_contig.fasta
│ ├── all_contig.fasta.fai
│ └── all_contig.fastq
├── per_sample_outs
│ └── beam-ab
│ ├── antigen_analysis
│ ├── count
│ ├── metrics_summary.csv
│ ├── vdj_t
│ └── web_summary.html
└── vdj_reference
├── fasta
│ ├── donor_regions.fa
│ └── regions.fa
└── reference.json