Cell Ranger7.1, printed on 01/31/2025
| After June 30, 2023, new Cell Ranger releases will no longer support Targeted Gene Expression analysis. |
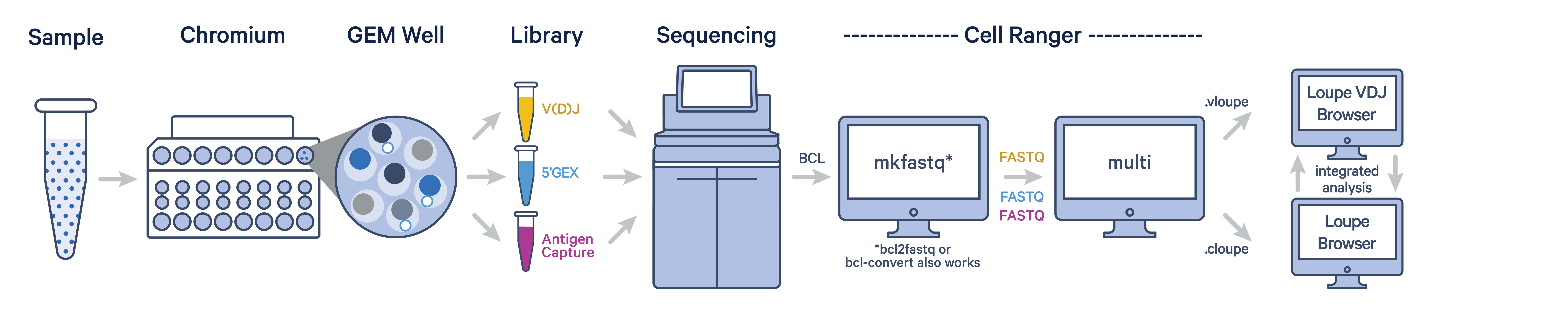
The Chromium Single Cell 5’ Barcode Enabled Antigen Mapping (BEAM) technology offers a scalable approach for mapping a V(D)J receptor to a target antigen by enabling the detection of gene expression profiles, paired V(D)J receptors, and signal from a bound antigen from the same single cell. All of these libraries, generated from a single GEM well, can be analyzed together with Cell Ranger v7.1 or later using the cellranger multi pipeline.
Hereafter libraries created using the BEAM-T workflow are called TCR Antigen Capture libraries, whereas libraries created using the BEAM-Ab workflow are called BCR Antigen Capture libraries. Antigen Capture without a prefix refers to either TCR or BCR Antigen Capture library, depending on the context.
To analyze an antigen library created using an antigen-multimer staining assay (TotalSeq™-C, Immudex's dMHC Dextramer® libraries with dCODE Dextramers), visit the TotalSeq™-C section of the 3' Chromium Single Cell Gene Expression with Feature Barcode technology page.
| Antigen Capture (BEAM) analysis is not supported on the 10x Genomics Cloud Platform. |
| Antibody Capture (cell surface protein) Feature Barcode libraries can also be generated and analyzed with Gene Expression, Antigen Capture, and V(D)J libraries. See the example config CSV section to include an Antibody Capture library in your analysis. |
| Antigen Capture and CRISPR Guide Capture cannot be performed in the same experiment. |
Antigen Capture + VDJ + Gene Expression (GEX) is the minimum set of libraries necessary to process antigen data from a single experiment. Here is a list of supported and unsupported library combinations. Please note that this table is not an exhaustive list.
| Library Combination | Supported? | TCR Antigen Capture + VDJ-T + GEX | ✅ | BCR Antigen Capture + VDJ-B + GEX | ✅ | Antigen Capture + VDJ + GEX + Antibody Capture | ✅ | TCR Antigen Capture + VDJ-T-GD + GEX | ❎ |
|---|---|
| Antigen Capture only | ❌ |
| Antigen Capture + VDJ | ❌ |
| TCR Antigen Capture + BCR Antigen Capture | ❌ |
| Antigen Capture + CRISPR Guide Capture | ❌ |
The following inputs are needed to analyze Antigen Capture, V(D)J, and Gene Expression libraries together using the cellranger multi pipeline:
Any Cell Ranger workflow starts by demultiplexing the Illumina sequencer's base call files (BCLs) for each flow cell directory into FASTQ files. Follow the instructions on running cellranger mkfastq, Illumina's BCL convert, or bcl2fastq to generate Gene Expression, V(D)J, and Antigen Capture FASTQ files. If you are already starting with FASTQ files, you can skip this step.
A Feature Reference CSV file is required when processing any Feature Barcode data. General details of this file are available on the 3' Single Cell Gene Expression website.
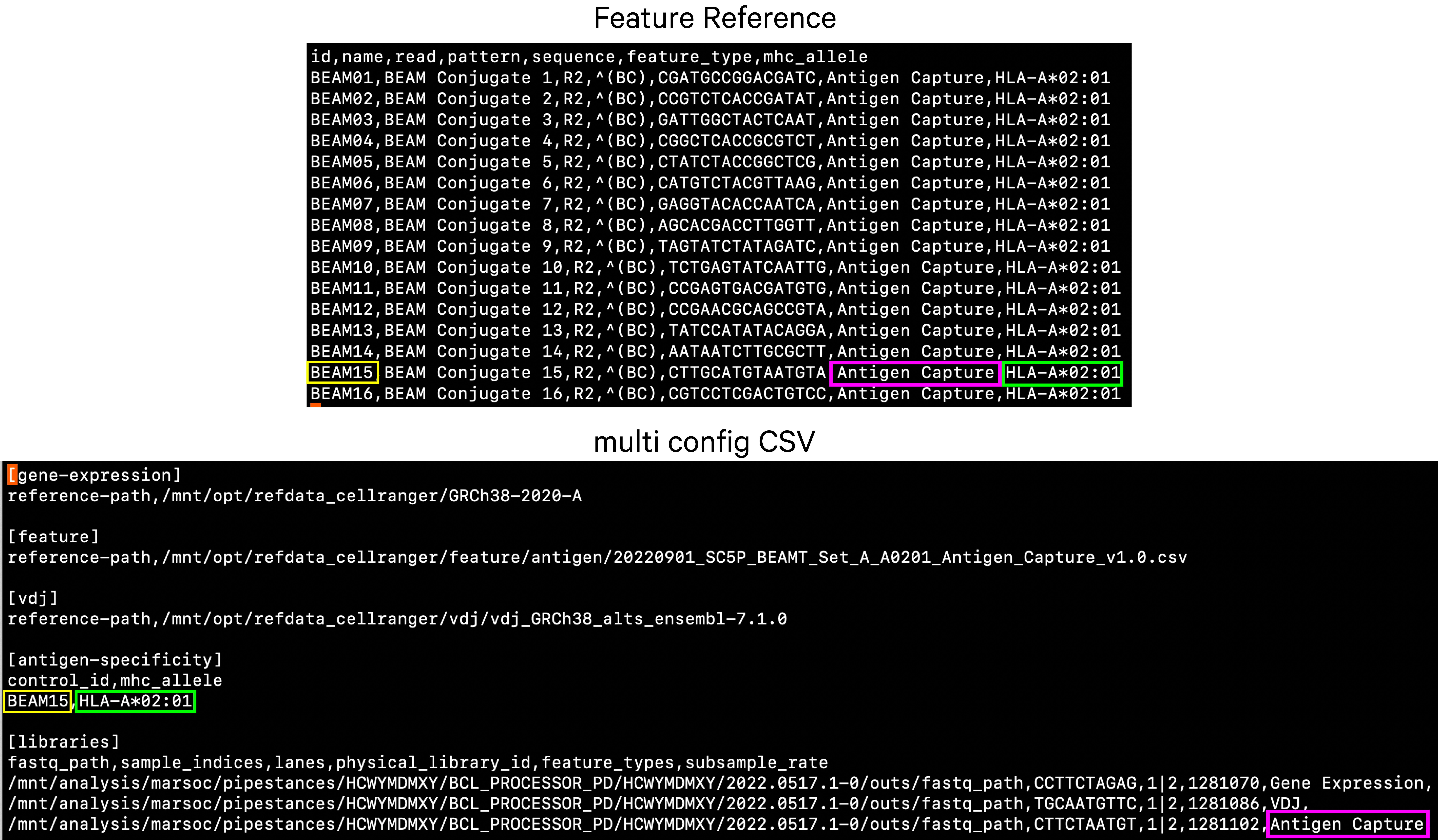
If a TCR Antigen Capture library is present, the Feature Reference must have an additional column, mhc_allele, that defines the MHC allele associated with each antigen included in the experiment.
id,name,read,pattern,sequence,feature_types,mhc_allele ag1,ag1,R2,^(BC),CGATGCCGGACGATC,Antigen Capture,A*01 ag2,ag2,R2,^(BC),CCGTCTCACCGATAT,Antigen Capture,A*01 neg_control01,neg_control01,R2,^(BC),GATTGGCTACTCAAT,Antigen Capture,A*01 ag4,ag4,R2,^(BC),CGGCTCACCGCGTCT,Antigen Capture,A*02 ag5,ag5,R2,^(BC),CTATCTACCGGCTCG,Antigen Capture,A*02 neg_control02,neg_control02,R2,^(BC),CATGTCTACGTTAAG,Antigen Capture,A*02 ab1,ab1,R2,^(BC),GAGGTACACCAATCA,Antibody Capture,A*01
If one Antigen Capture id has an entry in the mhc_allele column, all IDs must have an entry in that column. The mhc_allele name for the negative control peptide must match the name specified in the [antigen-specifity] section of the multi config CSV. Similarly, the id field for the control antigen in the Feature Reference CSV must match the control_id in the multi config CSV.
For BCR Antigen Capture, the mhc_allele column must NOT be included:
id,name,read,pattern,sequence,feature_types ag1,ag1,R2,^(BC),CGATGCCGGACGATC,Antigen Capture ag2,ag2,R2,^(BC),CCGTCTCACCGATAT,Antigen Capture ag4,ag4,R2,^(BC),CGGCTCACCGCGTCT,Antigen Capture ag5,ag5,R2,^(BC),CTATCTACCGGCTCG,Antigen Capture ag6,ag6,R2,^(BC),CATGTCTACGTTAAG,Antigen Capture ag7,ag7,R2,^(BC),GAGGTACACCAATCA,Antigen Capture ag8,ag8,R2,^(BC),AGCACGACCTTGGTT,Antigen Capture neg_control01,neg_control01,R2,^(BC),GATTGGCTACTCAAT,Antigen Capture
The id field for the control antigen in the Feature Reference CSV must match the control_id in the multi config CSV.
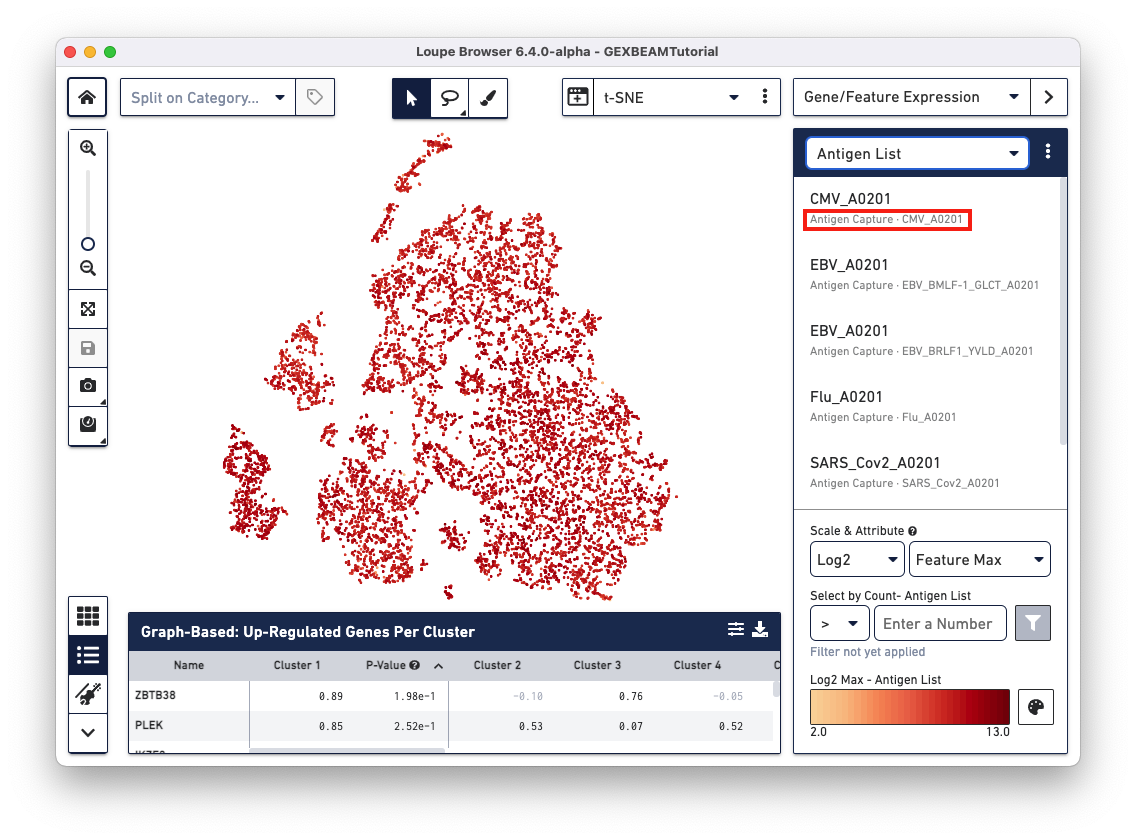
Loupe V(D)J Browser v5.0 and Loupe Browser v6.4 extract antigen names from the Feature Reference CSV. These names cannot be changed via the browser interface. In this Feature Reference example, the id and name fields have the same entry for each antigen. In Loupe Browser, the name field corresponds to the antigen name in grey (as shown below):
The Feature Barcode sequences of available BEAM conjugates section lists all the BEAM conjugate Feature Barcode sequences.
The cellranger multi pipeline requires a configuration CSV file as input, referred to here as the multi config CSV. The multi config CSV contains paths to FASTQ files for V(D)J, Gene Expression, and Antibody Capture (BEAM) libraries, as well as a path to the Feature Reference CSV.
The multi config CSV contains both the library definitions and experiment configuration variables, comprising up to five sections:
[gene-expression][feature][vdj][antigen-specificity][libraries]The [gene-expression], [feature], [antigen-specificity], and [vdj] sections have at most two columns and are responsible for configuring their respective portions of the experiment. The [libraries] section specifies where input FASTQ files may be found.
A customizable template for a multi config CSV can be downloaded here, and example multi config CSVs can be downloaded from public datasets. Cell Ranger v7.1 and later also provides the option to download a multi config CSV template via the command line.
Here are the example multi config CSVs for a few commonly used library combinations with Antigen Capture. Make sure to replace /path/to with the full path to your data, and edit any text in red according to the experiment's sample/library/file names.
Cell Ranger v7.1 enables users to download a multi config CSV template by running:
cellranger multi-template --output=/path/to/FILE.csv
Replace code in red with the path to a directory in which you wish to output the template. Omitting the file path downloads the file into your working directory. After downloading, remember to customize the template based on your assay and experimental design.
To print a list and description of all configurable parameters available in cellranger multi, run
cellranger multi-template --parameters
Specifying both --parameters and --output will output a parameter documentation file. Run cellranger multi-template --help or cellranger multi-template -h for more information about available flags.
All customizable parameters per section are described here:
| multi config CSV | |
|---|---|
Section: [gene-expression] | |
| Field | Description |
reference | Path of folder containing 10x Genomics-compatible reference. Required for Gene Expression and Feature Barcode libraries. |
target-panel | Optional. Path to a target panel CSV file or name of a 10x Genomics fixed gene panel (pathway, pan-cancer, immunology, neuroscience). |
no-target-umi-filter | Optional. Disable targeted UMI filtering stage. Default: false. |
r1-length | Optional. Limit the length of the input Read 1 sequence of Gene Expression libraries to the first N bases, where N is a user-supplied value. The length includes the Barcode and UMI sequences so do not set this below 26. This and r2-length are useful options for determining the optimal read length for sequencing. Default: do not trim Read 1. |
r2-length | Optional. Limit the length of the input Read 2 sequence of Gene Expression libraries to the first N bases, where N is a user-supplied value. Trimming occurs before sequencing metrics are computed and therefore, limiting the length of Read 2 may affect Q30 scores. Default: do not trim Read 2. |
chemistry | Optional. Assay configuration. NOTE: by default, the assay configuration is detected automatically, which is the recommended mode. In general, users will not need to specify a chemistry. Options are: auto for auto-detection, fiveprime for Single Cell 5', SC5P-PE for paired-end or SC5P-R2 for R2-only, SC-FB for Single Cell Antibody-only. Default: auto. |
expect-cells | Optional. Override the pipeline’s auto-estimate. See cell calling algorithm overview for details on how this parameter is used. If used, enter the expected number of recovered cells. |
force-cells | Optional. Force pipeline to use this number of cells, bypassing cell-calling algorithm. |
include-introns | Optional. Set to false to exclude intronic reads in count. Including introns in analysis is recommended to maximize sensitivity, except when target-panel is used. Default: true |
no-secondary | Optional. Disable secondary analysis, e.g., clustering. Default: false. |
no-bam | Optional. Set this flag to true to skip BAM file generation. This will reduce the total computation time for the pipestance and the size of the output directory. If unsure, we recommend not using this option, as BAM files can be useful for troubleshooting and downstream analysis. Default: false |
check-library-compatibility
| Optional. Allows users to disable the check that evaluates 10x Barcode overlap between libraries when multiple libraries are specified (e.g., Gene Expression + Antibody Capture). Setting this option to false will disable the check across all library combinations. We recommend running this check (default), however, if an error occurs, users can bypass the check to generate outputs for troubleshooting. Default: true |
Section: [feature] | |
| Field | Description |
reference | Required. Feature reference CSV file, declaring Antigen Capture (and Antibody Capture) constructs and associated barcodes. |
r1-length | Optional. Limit the length of the input Read 1 sequence of Feature Barcode libraries to the first N bases, where N is a user-supplied value. The length includes the Barcode and UMI sequences so do not set this below 26. This and r2-length are useful options for determining the optimal read length for sequencing. Default: do not trim Read 1. |
r2-length | Optional. Limit the length of the input Read 2 sequence of Feature Barcode libraries to the first N bases, where N is a user-supplied value. Trimming occurs before sequencing metrics are computed and therefore, limiting the length of Read 2 may affect Q30 scores. Default: do not trim Read 2. |
Section: [vdj] | |
| Field | Description |
reference | Path of folder containing 10x Genomics-compatible V(D)J reference. Required for V(D)J Immune Profiling libraries. |
inner-enrichment-primers | Optional. If inner enrichment primers other than those provided in the 10x Genomics kits are used, they need to be specified here as a text file with one primer per line. |
r1-length | Optional. Limit the length of the input Read 1 sequence of V(D)J libraries to the first N bases, where N is a user-supplied value. The length includes the Barcode and UMI sequences so do not set this below 26. This and r2-length are useful options for determining the optimal read length for sequencing. Default: do not trim Read 1. |
r2-length | Optional. Limit the length of the input Read 2 sequence of V(D)J libraries to the first N bases, where N is a user-supplied value. Trimming occurs before sequencing metrics are computed and therefore, limiting the length of Read 2 may affect Q30 scores. Default: do not trim Read 2. |
Section: [libraries] | |
| Column | Description |
fastq_id | Required. The Illumina sample name to analyze. This will be as specified in the sample sheet supplied to mkfastq or bcl2fastq. |
fastqs | Required. The folder containing the FASTQ files to be analyzed. Generally, this will be the fastq_path folder generated by cellranger mkfastq. |
lanes | Optional. The lanes associated with this sample, separated by |. Defaults to using all lanes. |
feature_types | Required. The underlying feature type of the library, which must be one of Gene Expression, VDJ, VDJ-T, VDJ-T-GD, VDJ-B, Antibody Capture, or Antigen Capture. Please note that Antigen Capture should be used only for BEAM libraries. For other (non-BEAM) antigen libraries (TotalSeq™-C, Immudex's dMHC Dextramer® libraries with dCODE Dextramers), set feature_types to Antibody Capture. Learn about feature_types auto-detection. |
subsample_rate | Optional. The rate at which reads from the provided FASTQ files are sampled. Must be strictly greater than 0 and less than or equal to 1. |
Section: [antigen-specificity] Recommended if Antigen Capture (BEAM) library present. Needed for antigen specificity score calculation. | |
| Field | Description |
control_id | Required. A user-defined ID for any negative controls used in the T/BCR Antigen Capture assay. Must match id specified in the Feature Reference CSV. May only include ASCII characters and must not use whitespace, slash, quote, or comma characters. Each ID must be unique and must not collide with a gene identifier from the transcriptome. |
mhc_allele | The MHC allele for TCR Antigen Capture libraries. Must match mhc_allele name specified in the Feature Reference CSV. For BCR Antigen Capture library, analysis runs with or without this header. If you keep the header, leave rows blank. |
For help on how to configure the [libraries] section to target a particular set of FASTQs, consult Specifying Input FASTQ Files for cellranger multi.
|
| BEAM Conjugate | Sequence | BEAM Conjugate 1 | CGATGCCGGACGATC | BEAM Conjugate 2 | CCGTCTCACCGATAT | BEAM Conjugate 3 | GATTGGCTACTCAAT | BEAM Conjugate 4 | CGGCTCACCGCGTCT | BEAM Conjugate 5 | CTATCTACCGGCTCG | BEAM Conjugate 6 | CATGTCTACGTTAAG | BEAM Conjugate 7 | GAGGTACACCAATCA | BEAM Conjugate 8 | AGCACGACCTTGGTT | BEAM Conjugate 9 | TAGTATCTATAGATC | BEAM Conjugate 10 | TCTGAGTATCAATTG | BEAM Conjugate 11 | CCGAGTGACGATGTG | BEAM Conjugate 12 | CCGAACGCAGCCGTA | BEAM Conjugate 13 | TATCCATATACAGGA | BEAM Conjugate 14 | AATAATCTTGCGCTT | BEAM Conjugate 15 | CTTGCATGTAATGTA | BEAM Conjugate 16 | CGTCCTCGACTGTCC |
|---|
These images highlight the relationship between a TCR Antigen Capture Feature Reference CSV and its multi config CSV:
The cellranger multi pipeline runs with two required arguments:
| Argument | Description |
|---|---|
--id | A unique run ID string: e.g. sample345 that is also the output folder name. Cannot be more than 64 characters. |
--csv | Path to multi config CSV file enumerating input libraries and analysis parameters. |
--description | Optional. Sample description to embed in output files. |
After obtaining the necessary inputs, and determining the input arguments, it is time to run cellranger multi. Remember to replace the bits of code in red with your sample id and csv file path:
mkdir /home/jdoe/runs cd /home/jdoe/runs cellranger multi --id=sample345 --csv=/home/jdoe/sample345.csv
Learn more about a successful cellranger multi run on the cellranger multi pipelines page, or learn about the expected output file structure under the Understanding Outputs section.
antigen_analysis/ folder located inside per_sample_outs/.